
Kohn–Sham密度泛函理论(KSDFT)是化学、物理和材料科学中应用最广泛的电子结构方法,这种普遍性源于KSDFT的良好精度与成本平衡。尽管如此,研究人员在大型复杂分子系统的预测模拟中使用KSDFT方法面临着计算成本的挑战。在第一性原理分子动力学的背景下,挑战是与Kohn–Sham轨道计算相关的高成本因素。近日,吉林大学Wenhui Mi、南京理工大学Luo Kai、佛罗里达大学S. B. Trickey、罗格斯大学Michele Pavanello综述研究了无轨道密度泛函理论。
本文要点:
1) 顾名思义,无轨道DFT完全避开了这些轨道的明确使用。如果没有它们,KSDFT的结构和算法复杂性将大大简化,并且无论系统状态如何,都可以实现随系统大小的近似线性缩放。因此,与传统的KSDFT相比,更大的系统尺寸和更长的模拟时间尺度将成为可能,从而可以探索新的化学现象和新的材料。在这篇综述中,作者介绍了OFFT的历史背景、理论基础,以及通过近似动能密度泛函(KEDF)实现中的挑战。
2) 作者回顾了一系列KEDF在这一挑战方面的最新进展,如一点、两点和机器学习,以及一些探索较少的形式。作者强调使用精确的约束和设计选择的必然性。然后,作者研究了相关的数值技术,并实现了OFFT特有的算法。最后,作者展示了OFFT在材料科学、化学和物理领域的强大应用。
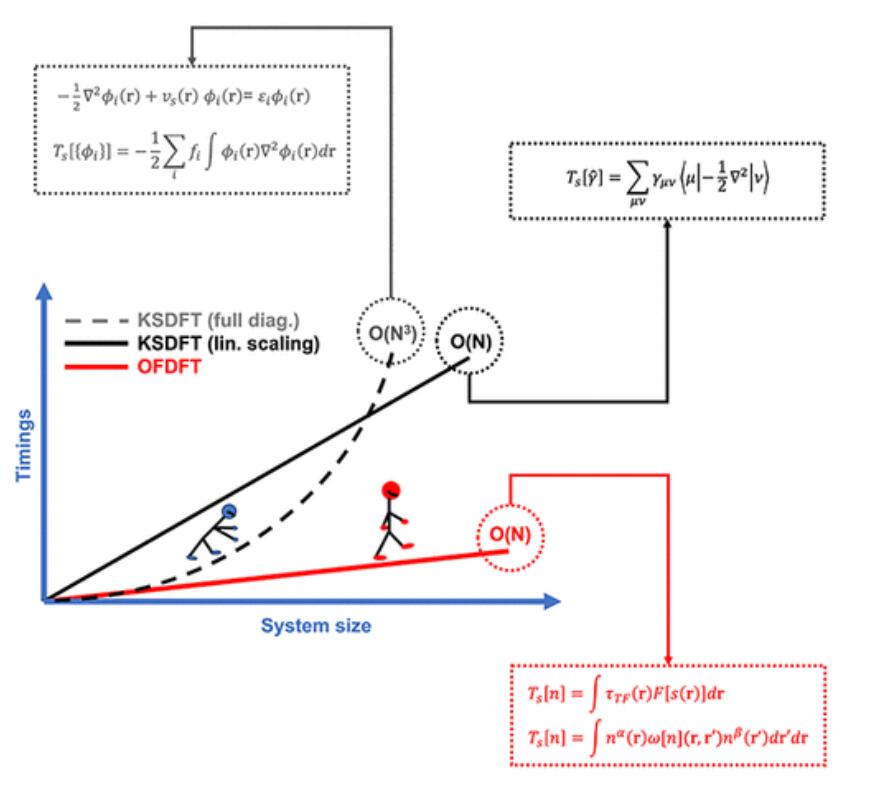
Wenhui Mi et.al Orbital-Free Density Functional Theory: An Attractive Electronic Structure Method for Large-Scale First-Principles Simulations Chem. Rev. 2023
DOI: 10.1021/acs.chemrev.2c00758
https://doi.org/10.1021/acs.chemrev.2c00758

















