 Fe-N-C(铁-氮-碳)电催化剂已成为用于氧还原反应(ORR)的贵金属基材料的有效替代品。然而,这些材料在电化学条件下的结构尚不清楚,并且它们在酸性环境中的较差稳定性对成功应用于商业燃料电池构成了巨大挑战。为了对这些复杂现象提供分子水平的见解,普渡大学Jeffrey Greeley结合了周期密度泛函理论(DFT)计算、对ORR反应中间体(包括O和OH)的共吸附效应进行了详尽分析,以及溶剂化稳定效应的综合分析,以构建描述活性位点原位结构的电压相关从头算热力学相图。
Fe-N-C(铁-氮-碳)电催化剂已成为用于氧还原反应(ORR)的贵金属基材料的有效替代品。然而,这些材料在电化学条件下的结构尚不清楚,并且它们在酸性环境中的较差稳定性对成功应用于商业燃料电池构成了巨大挑战。为了对这些复杂现象提供分子水平的见解,普渡大学Jeffrey Greeley结合了周期密度泛函理论(DFT)计算、对ORR反应中间体(包括O和OH)的共吸附效应进行了详尽分析,以及溶剂化稳定效应的综合分析,以构建描述活性位点原位结构的电压相关从头算热力学相图。
本文要点:
1) 这些结构进一步与活性和稳定性描述符联系在一起,这些描述符可以与实验参数进行比较,例如ORR的半波电位和碳腐蚀和CO2析出的起始电位。结果表明,与本体中的位点相比,锯齿形碳边缘处的吡啶基Fe位点以及其他边缘位点对ORR表现出高活性。
2) 然而,与活性位点相邻的边缘易于通过过氧化和随后的位点损失而不稳定。结果表明,合成具有小尺寸和大边缘长度的Fe-N-C催化剂可以提高ORR活性,而在燃料电池操作过程中应限制电压波动以防止过度氧化边缘的碳腐蚀。
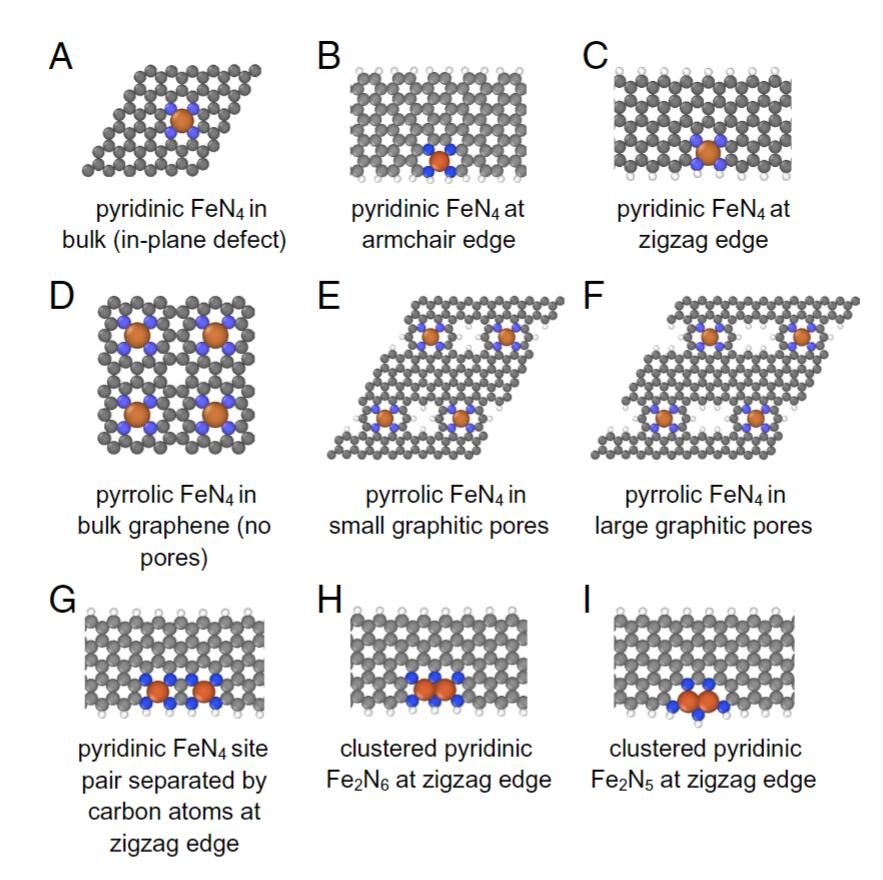
Ankita Morankar et.al A first principles analysis of potential-dependent structural evolution of active sites in Fe-N-C catalysts PNAS 2023
DOI: 10.1073/pnas.2308458120
https://doi.org/10.1073/pnas.2308458120

















