 由于可再生能源的采用,对高安全性、高性能和低成本储能系统(EES)的需求不断增加,正逐渐超过商用锂离子电池(LIBs)的能力。固态电解质(SSE),包括无机物、聚合物和复合材料,已成为极具潜力的下一代全固态电池(ASSB)。ASSB提供了更高的理论能量密度、改进的安全性和扩展的循环稳定性,使其在学术界和工业界越来越受关注。然而,ASSB的商业化仍然面临着重大挑战,如高界面电阻和枝晶快速生长。为了克服这些问题,深入了解SSE材料复杂的化学-电化学-机械相互作用至关重要。近日,南京工业大学Zhu Yusong、Zheng Zhuoyuan综述研究了锂二次电池固态电解质中的计算方法。
由于可再生能源的采用,对高安全性、高性能和低成本储能系统(EES)的需求不断增加,正逐渐超过商用锂离子电池(LIBs)的能力。固态电解质(SSE),包括无机物、聚合物和复合材料,已成为极具潜力的下一代全固态电池(ASSB)。ASSB提供了更高的理论能量密度、改进的安全性和扩展的循环稳定性,使其在学术界和工业界越来越受关注。然而,ASSB的商业化仍然面临着重大挑战,如高界面电阻和枝晶快速生长。为了克服这些问题,深入了解SSE材料复杂的化学-电化学-机械相互作用至关重要。近日,南京工业大学Zhu Yusong、Zheng Zhuoyuan综述研究了锂二次电池固态电解质中的计算方法。
本文要点:
1) 计算方法在揭示SSE相关基本机制和加速其发展方面发挥了至关重要的作用,从原子第一性原理计算、分子动力学模拟、多物理建模到机器学习方法。这些方法能够预测固有特性和界面稳定性,并研究材料退化,探索拓扑设计等因素。
2) 作者概述了SSE研究中使用的不同数值方法,并讨论了数值辅助方法的知识现状,特别关注机器学习方法,以了解SSE在不同空间和时间尺度上的多物理耦合。此外,作者还强调了SSE进步的见解和前景。
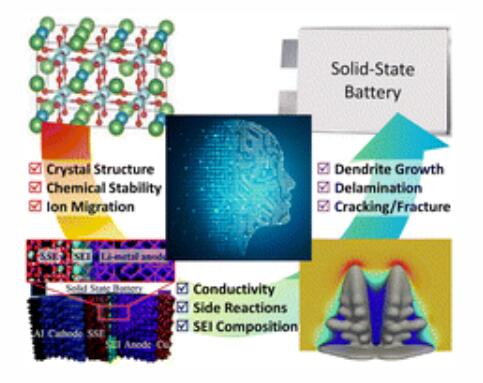
Zhuoyuan Zheng et.al Computational approach inspired advancements of solid-state electrolytes for lithium secondary batteries: from first-principles to machine learning Chem. Soc. Rev. 2024
DOI: 10.1039/D3CS00572K
https://doi.org/10.1039/D3CS00572K

















