
如何处理连续弯曲的分子是从低温电子显微镜(cryo-EM)图像中分析蛋白质单粒子的最大突出挑战之一。鉴于此,来自剑桥大学MRC分子生物实验室的Sjors H. W. Scheres、Johannes Schwab等人开发了一种软件工具DynaMight。
文章要点:
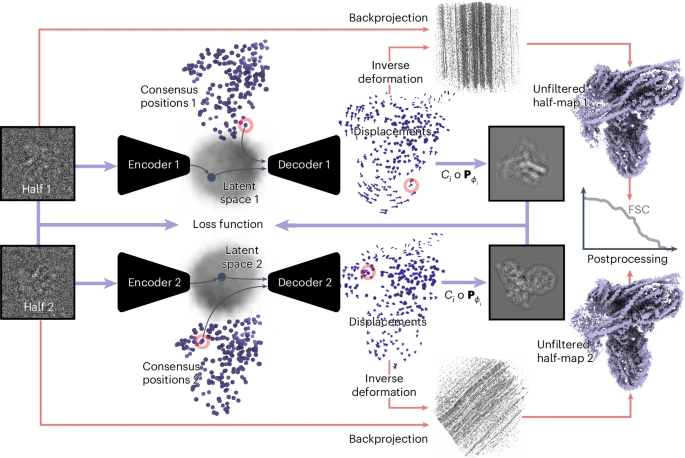
1) 该研究开发的这种工具是通过学习每个粒子图像的共识结构的高斯伪原子模型的三维变形,以此来估计低温EM数据集中的连续构象空间;
2) 此外,该研究还还展示了如何通过在半组冷冻EM数据上独立训练两个变分自编码器来获得变形的误差估计,以及通过使用原子模型对三维变形进行正则化可能会因模型偏差而导致重要伪影,DynaMight作为RELION-5的一部分作为免费开源软件分发。
参考资料:
Schwab, J., Kimanius, D., Burt, A. et al. DynaMight: estimating molecular motions with improved reconstruction from cryo-EM images. Nat Methods (2024).
10.1038/s41592-024-02377-5
https://doi.org/10.1038/s41592-024-02377-5

















